


背景介绍
稠合环丁烷骨架是天然产物和药物分子中的重要结构单元,但由于环张力及立体控制等挑战,其催化不对称合成方法仍十分有限。自1995年陆熙炎课题组开创性工作以来,膦催化的联烯酸酯环化反应已成为构建手性环状化合物的重要策略。联烯酸酯可作为C2、C3、C4和C6合成子参与多种环化反应,但迄今为止主要局限于构建普通或中等尺寸环系。近年来,中环化合物的跨环环化策略为构建复杂小环体系提供了新思路。
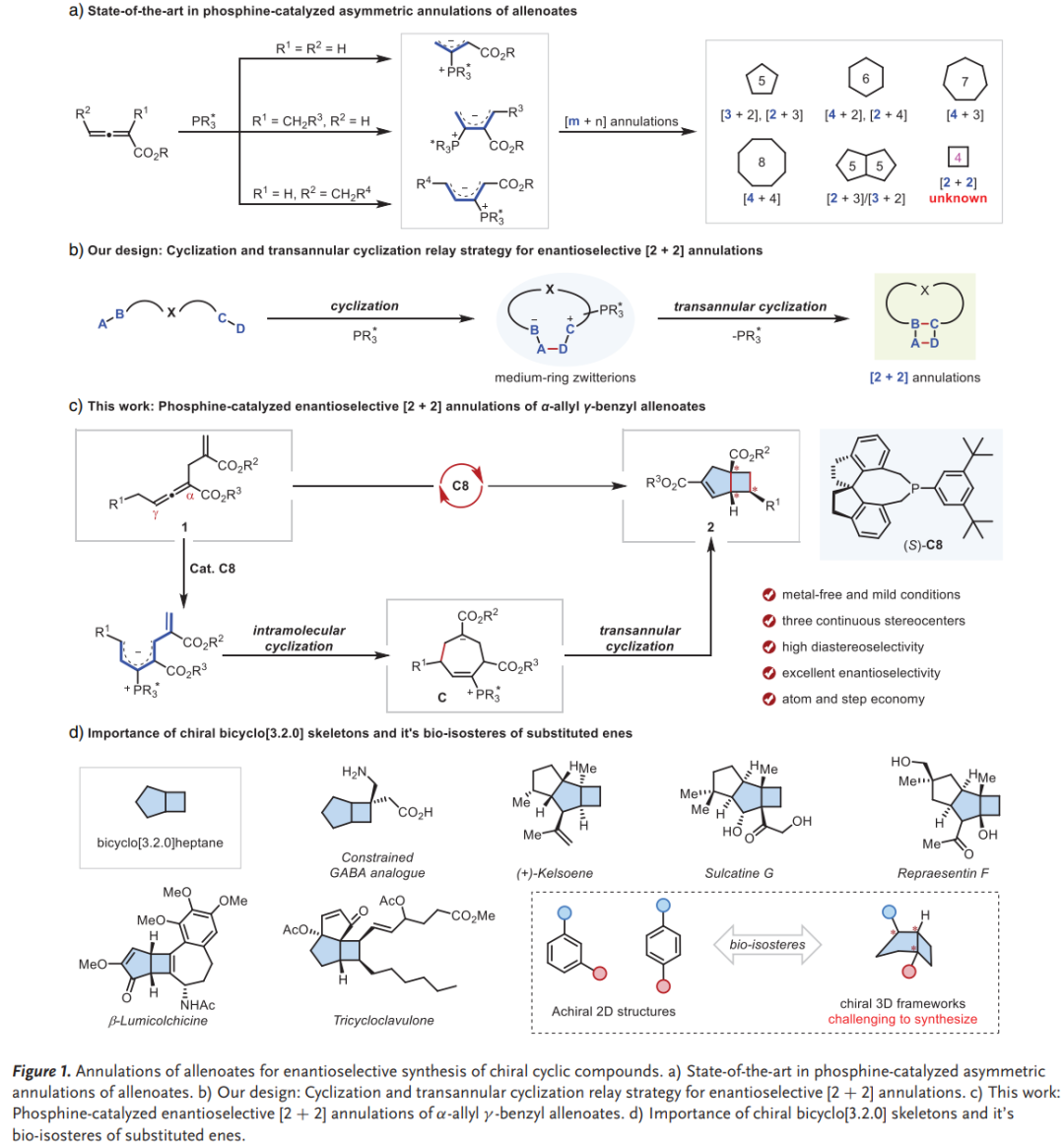
研究思路
本研究设计了一种创新策略:通过设计同时含有α-烯丙基和γ-苄基的联烯酸酯,利用其互补的亲电和亲核反应性,首先形成七元鏻盐中间体,随后发生跨环环化,高效构建手性双环[3.2.0]庚烯骨架。该策略的核心在于开发能够生成中环鏻盐并保留足够反应性进行后续跨环环化的催化体系。相关成果以“Phosphine-Catalyzed Enantioselective [2+2] Annulations of α-Allyl γ-Benzyl Allenoates via a Transannular Cyclization Approach”为题在《Angewandte Chemie International Edition》期刊上发表,南开大学化学学院黄有教授、肖力军研究员为论文通讯作者,南开大学有机新物质创造前沿科学中心为论文通讯单位。
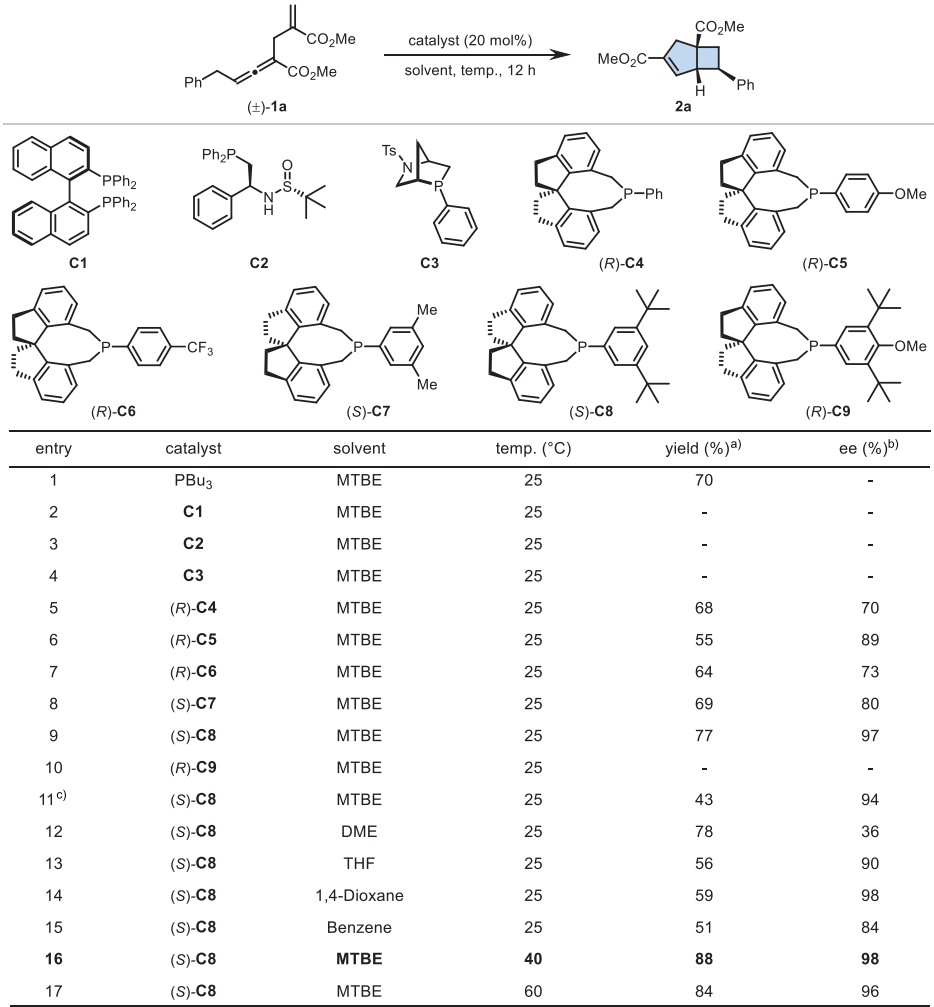
条件优化
以α-烯丙基γ-苄基联烯酸酯1a为模型底物进行条件优化。初步实验表明,PBu3在室温下能以70%收率得到外消旋产物2a。随后筛选多种手性膦催化剂发现,螺环膦催化剂C4能获得68%收率和70% ee。进一步优化显示,含有3,5-二叔丁基苯基的催化剂C8效果最佳,在40°C下能以88%收率和98% ee获得产物2a。
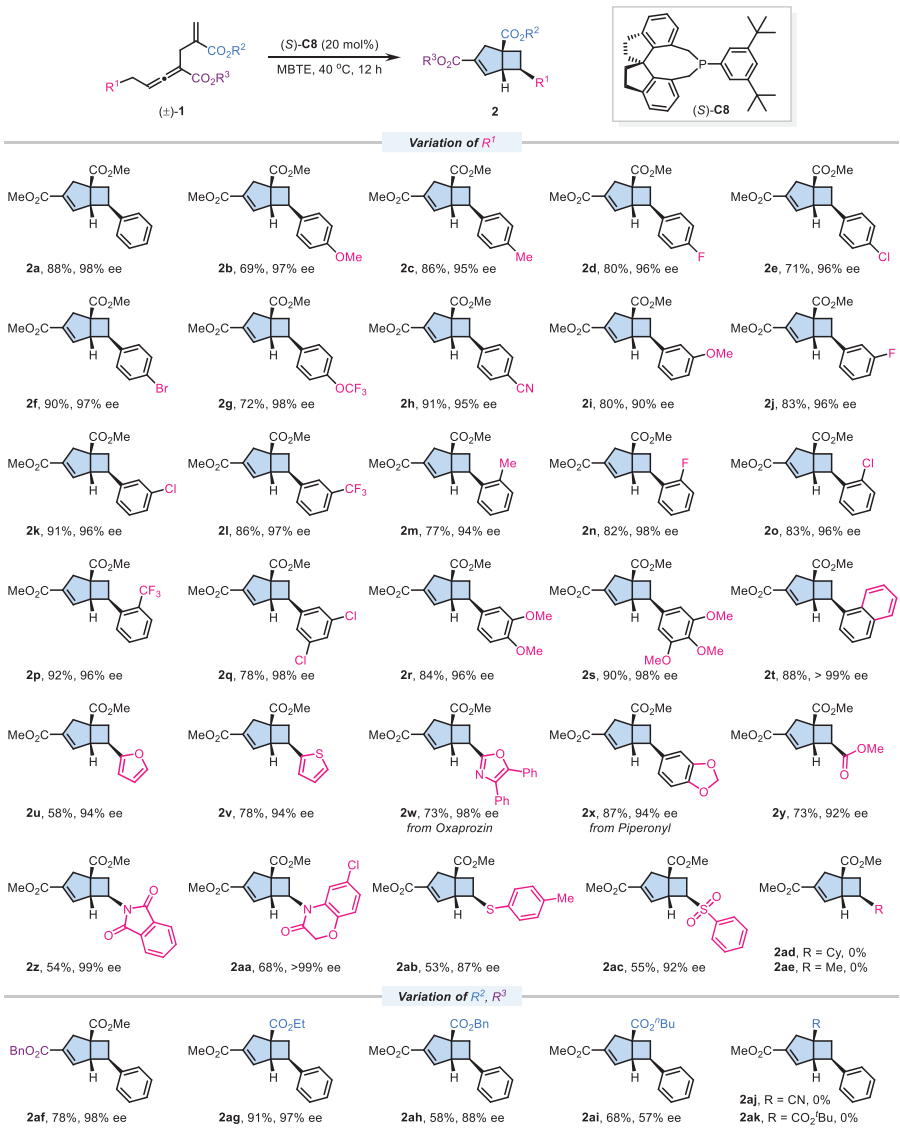
底物拓展
在最优条件下,该反应展现出广泛的底物适应性。多种对位、间位和邻位取代的苯基底物均能顺利反应,以良好收率(69-92%)和优异对映选择性(90-98% ee)得到相应产物。二取代或三取代苯基、萘基、呋喃和噻吩等杂芳基取代的底物也能良好兼容。特别值得注意的是,含有酯基、酰胺基和磺酰基的底物同样适用于该反应,展现了优异的官能团耐受性。
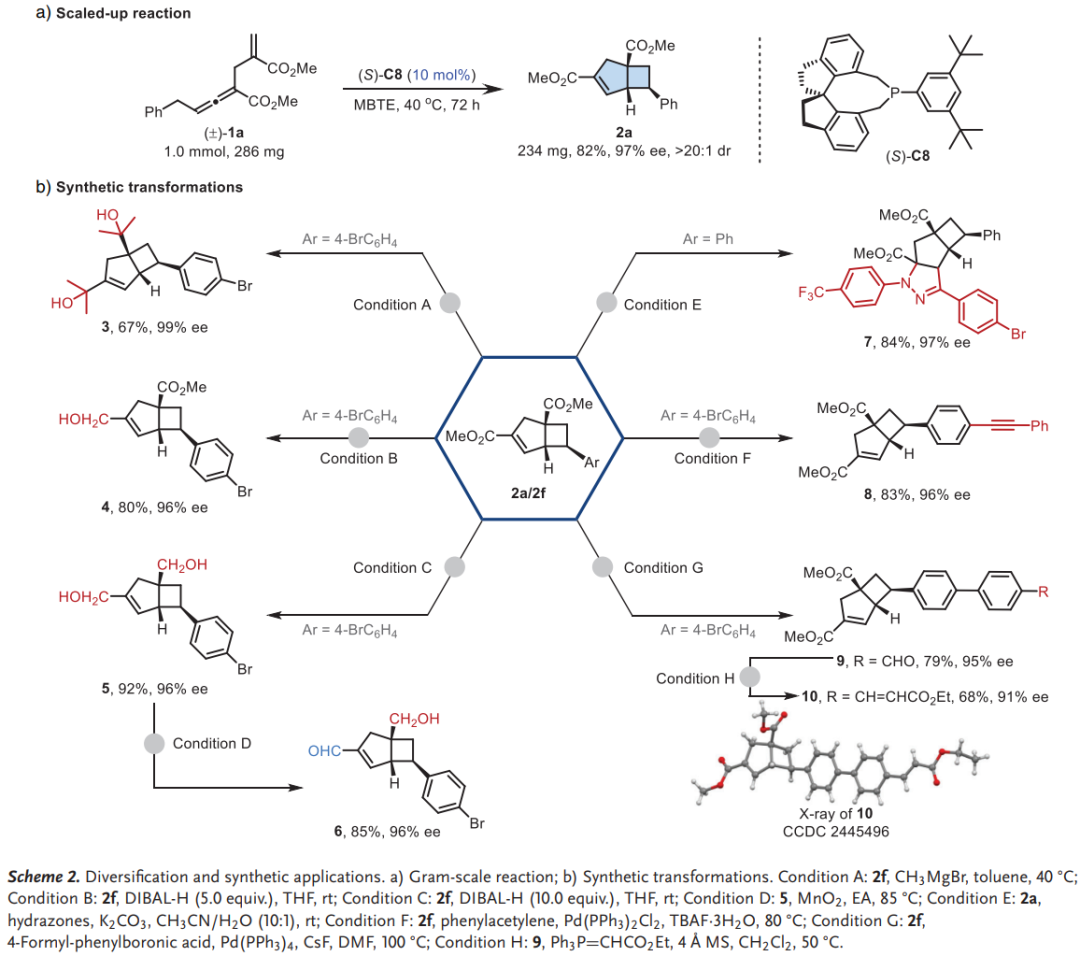
应用研究
克级规模实验证实了该方法的实用性,1.0 mmol规模反应仍能以82%收率和97% ee获得产物。产物可进行多样化衍生化,包括格氏试剂加成、DIBAL-H还原、[3+2]环加成以及Sonogashira和Suzuki偶联等反应,且对映纯度在整个转化过程中得以保持。通过X射线衍射分析确定了产物10的绝对构型,为其他产物的立体化学归属提供了依据。
机理研究
氘代实验表明反应中形成了碳负离子中间体。交叉实验证实了反应遵循分子内历程。中间体捕获实验成功分离到亲核加成产物15,支持七元环中间体的存在。时间进程实验显示产物ee值始终保持高位(96-98%),而回收底物的ee值逐渐增加至87%,表明存在动态动力学拆分过程。
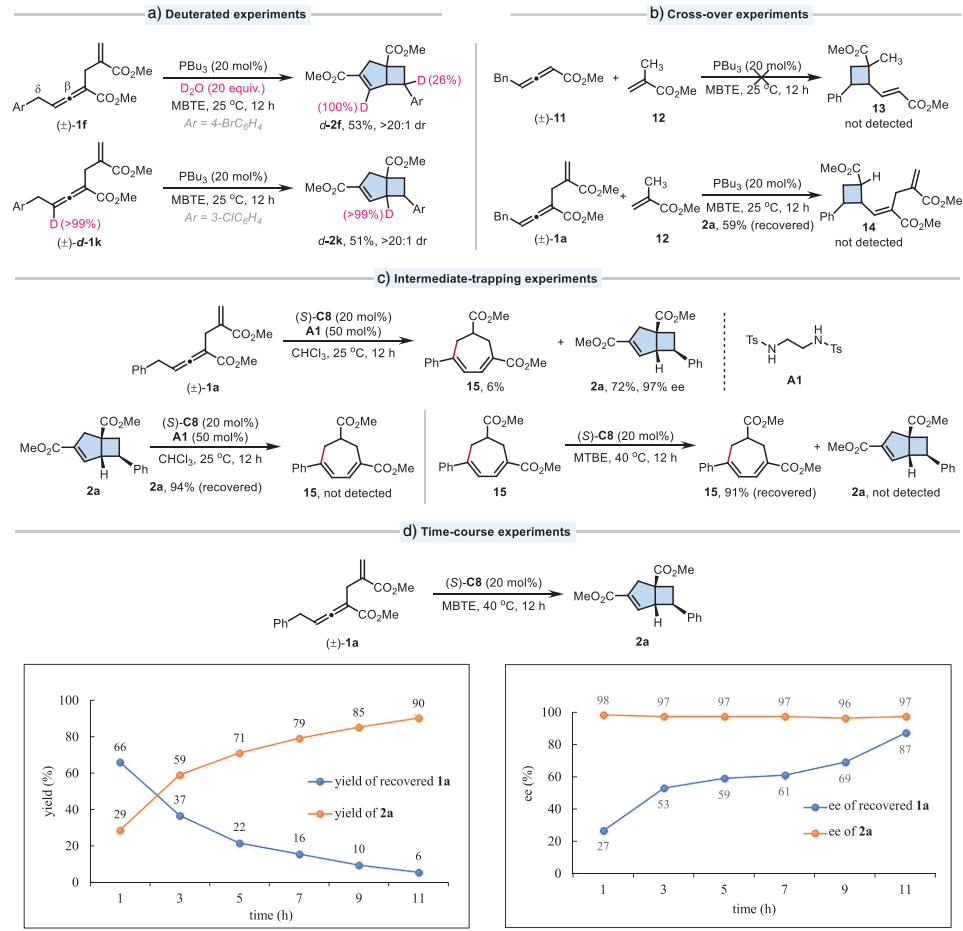
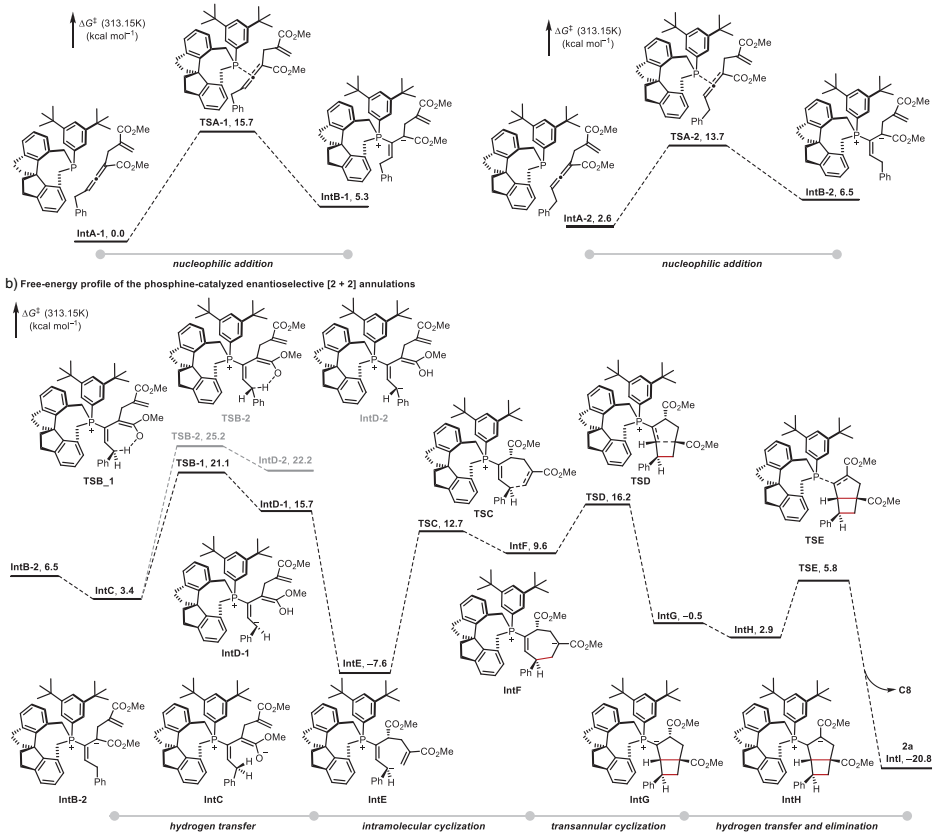
DFT计算揭示了详细反应机理:手性膦催化剂对联烯酸酯的亲核加成是可逆步骤,实现动态动力学拆分;δ-位碳的分子内氢转移去质子化是速率和对映选择性决定步骤;随后经历迈克尔加成形成七元环中间体,最后通过跨环环化生成最终产物。
总结
本研究发展了一种螺环膦催化的对映选择性[2+2]环化反应,通过跨环环化接力策略,实现了外消旋α-烯丙基γ-苄基联烯酸酯的立体汇聚式转化,为手性稠合环丁烷的高效合成提供了新方法。该方法具有条件温和、立体选择性高、底物范围广等优点,在天然产物和药物分子合成中具有重要应用价值。
文献详情
Xu-Ling Pan, Wei-Guo Xiao, Li-Jun Xiao, Qi-Lin Zhou, You Huang; Phosphine-Catalyzed Enantioselective[2+2] Annulations of α-Allyl γ-Benzyl Allenoates via a Transannular Cyclization Approach; Angewandte Chemie International Edition ; 10.1002/anie.202515642
来源:科学纵横
