


背景介绍
在有机合成中,1,4-二烯是一类非常重要的结构单元,广泛存在于天然产物和药物分子中(图1a)。合成这类结构的一个经典策略是利用烯丙基金属试剂与端炔反应。然而,由于端炔与烯丙基金属试剂的高反应活性,这类反应通常可在无催化条件下进行,其区域选择性由底物电子效应主导,符合电性匹配原则,几乎所有端炔的烯丙基金属化反应均表现出马氏选择性(即烯丙基加成至端炔的内侧碳,金属加成至外侧碳),生成支链1,4-二烯产物。目前尚未实现普通端炔反马氏烯丙基金属化的有效方法,这极大限制了产物的结构多样性,也制约了该类反应的实际应用。因此,开发一种可实现普通端炔反马氏烯丙基金属化的新型催化体系,作为对现有方法的补充,具有重要的合成价值。然而要实现这类反应,除需克服底物电子效应和背景反应的影响外,还需解决诸多问题:烯丙基金属试剂具有强碱性,极易与端炔的酸性C-H键反应生成炔基金属物种,进一步增强炔烃的电子极化,使区域选择性反转的难度大幅增加;此外,还需抑制炔烃多聚反应,并精准控制烯丙基加成的迁移选择性(图1b)。

图1. 研究背景
研究内容
近日,南开大学化学学院、有机新物质创造前沿科学中心朱守非教授课题组成功突破了这一限制,首次将2-亚氨喹啉铁配合物用于碳金属化反应,实现了端炔(包括芳基和烷基取代端炔)的反马氏烯丙基锌化反应。该方法能以高收率,立体和区域选择性制备以往难以获得的1,4-二烯基锌试剂,并已应用于多取代1,4-二烯以及多种天然产物关键中间体的合成(图1c)。
反应条件优化
作者首先以4-苯基苯乙炔(1aa)为模型底物,通过条件筛选最终获得最优条件:在2,6-二甲基取代的2-亚胺喹啉双齿配体铁配合物C7(2 mol%)的催化下,端炔1aa和烯丙基锌试剂(1.2 equiv)能以97%的收率和> 95:5的区域及立体选择性得到反马氏产物2aa(表1)。
表1. 反应条件优化

底物范围
该反应具有良好的官能团耐受性和底物普适性。如图2所示,无论是芳基还是烯基端炔,均能以优异的收率和选择性获得目标产物。此外,取代烯丙基锌试剂也能适用于该体系,得到非迁移烯丙基锌化的产物。
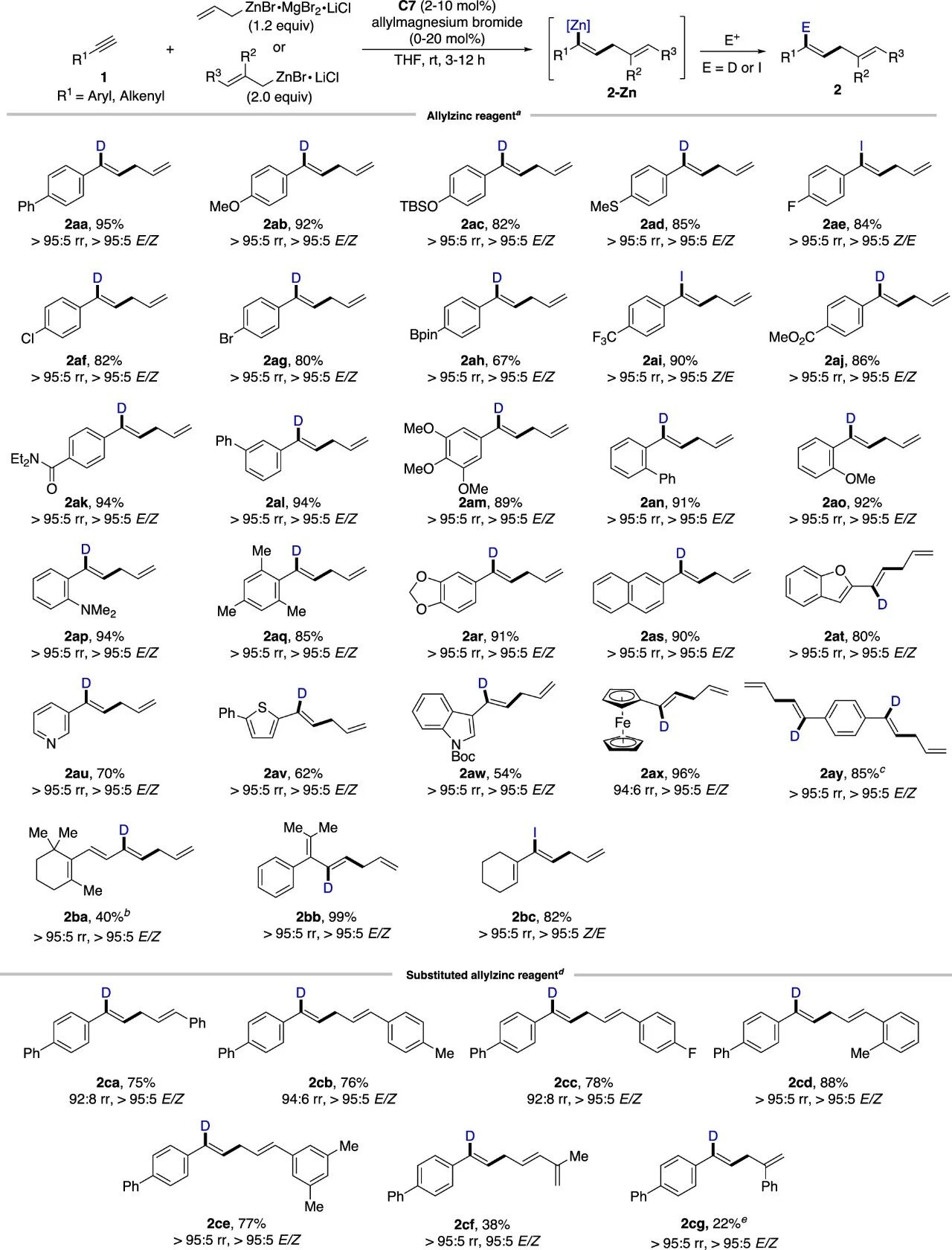
图2.芳基端炔底物范围
在更大位阻的2,6-二异丙基取代2-亚氨喹啉铁配合物C8的催化下,作者也实现了烷基端炔的烯丙基锌化反应。一级、二级和三级烷基端炔均适用于该体系,以≥ 93:7的区域选择性,> 95:5立体选择性得到反马氏烯丙基锌化产物(图3)。

图3.烷基端炔底物范围
产物转化与应用
该方法合成的新型1,4-二烯基锌试剂可与多种亲电试剂发生取代、加成、偶联等反应,高效构建复杂的多取代1,4-二烯化合物。与传统的氢烯丙基化、烯丙基卤化方法相比,该反应的产物具有更多的后期修饰途径,在合成化学中展现出显著优势。此外,该方法在一些含1,4-二烯骨架的功能分子合成中也具有良好的应用潜力。作者以此实现了RNA聚合酶抑制剂ripostatin B和瓜实蝇信息素的关键中间体的合成,合成效率显著优于文献报道的路线(图4)。

图4. 产物转化和合成应用
机理实验
为探究反应机理,作者开展了控制实验和计算研究。首先,作者通过当量还原实验说明了二价铁催化剂在体系中会被还原为零价(图5a)。接着作者成功制备了具有明确单晶结构的二烯配位零价铁配合物C8a(图5b)。该配合物可高效催化端炔1de与烯丙基锌试剂的反应:催化剂负载量仅为0.05 mol%时,反应可实现克级规模转化,转化数高达1900(图5c)。同时,作者发现C8a与烯丙基锌试剂以1:1比例混合时,体系颜色以及核磁氢谱信号发生了显著改变,同时作者也观察到二烯配体的解离。结合文献中相关低价铁活性物种的研究,作者认为反应生成的零价铁物种能与烯丙基锌试剂结合生成阴离子烯丙基铁物种,并参与整个催化循环(图5d)。

图5. 机理实验
DFT计算及可能的催化循环
为了进一步验证该种假设,以及探究反马氏选择性产生的原因,作者进行了DFT计算。结果表明,反应在三重态势能面上的能量显著低于单重态,其中反马氏迁移插入步骤(3Int1-C8 → 3Int2-C8 → 3TS1-C8 → 3Int1-C8)的总能垒为19.6 kcal/mol,表明该过程可在室温下顺利进行。相比之下,马氏反应路径总能垒为21.4 kcal/mol,比反马氏路径高1.8 kcal/mol,这与实验结果高度吻合(图6a)。
为探究2-亚胺喹啉铁配合物为何能实现这一特殊选择性,作者对三重态下的两个过渡态进行了弱相互作用、形变能/相互作用能、能量分解等分析。结果显示:由于2-亚氨喹啉配体两侧的空间位阻的差异,炔烃配位和迁移插入的空间取向会导致铁催化剂发生不同程度的形变:与非优势过渡态3TS1-r-C8相比,优势过渡态3TS1-C8的总形变能更低、总相互作用能更高;但形变能的影响(ΔΔEdist = 3.2 kcal/mol)要大于相互作用能的影响(ΔΔEint = −1.7 kcal/mol),对反应活化能做出了主要的贡献。从空间结构来看,3TS1-C8和3TS1-r-C8中亚胺氮原子所连芳环平面与亚胺双键所在平面之间的二面角θ分别为67.6°和62.7°,而低能中间体3Int1-C8中对应的二面角为76.6°。这一角度变化体现了催化剂的形变程度,与形变能的计算结果一致。以上结果说明2-亚胺喹啉配体合适的位阻是该选择性得以实现的关键(图6c)。
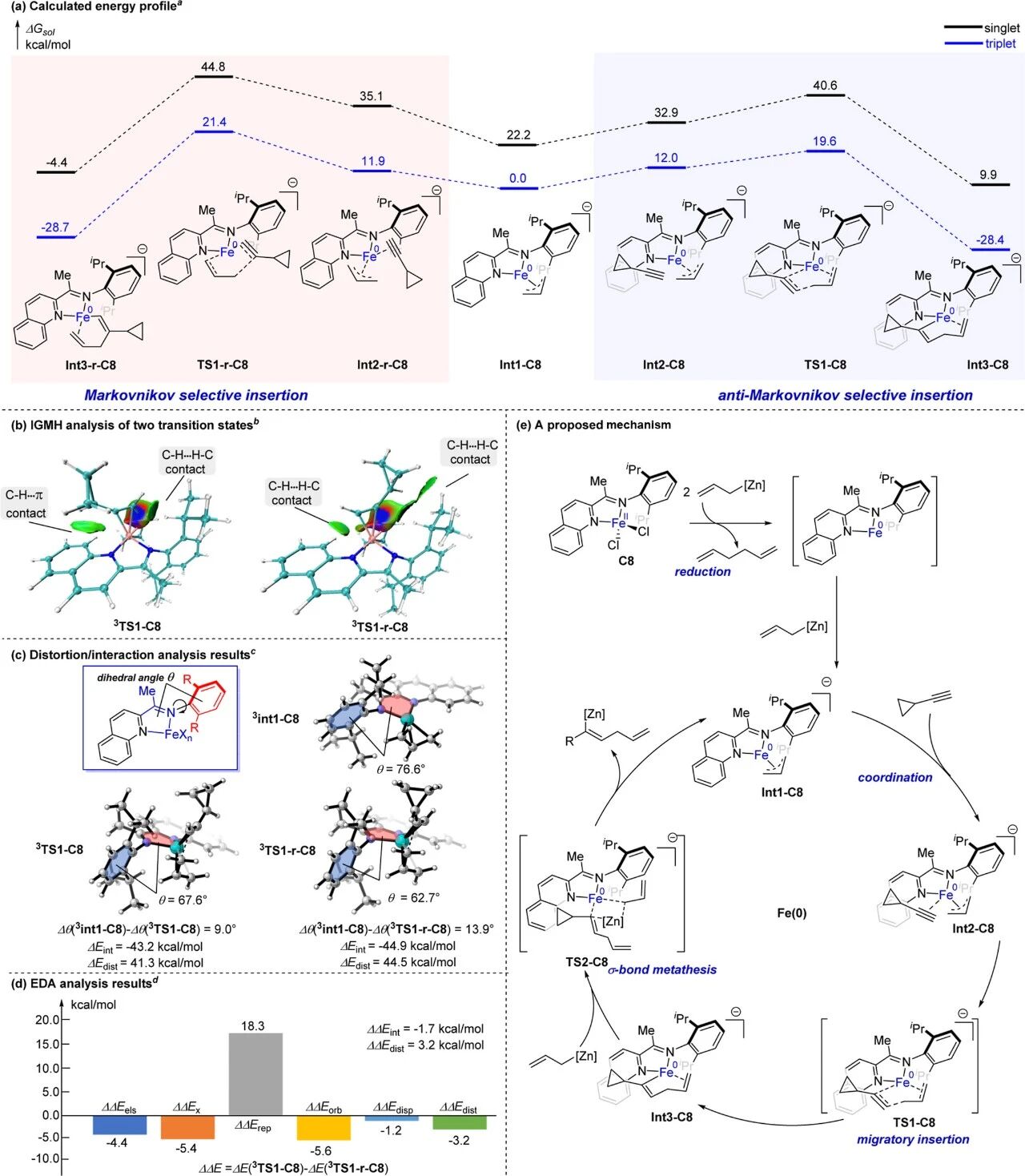
图6. DFT计算及可能的催化循环
基于上述实验和计算结果,作者提出了完整的催化循环:二价铁配合物C8首先被烯丙基锌试剂还原为零价,随后再与烯丙基锌试剂反应,生成阴离子烯丙基铁物种Int1-C8;Int1-C8与端炔配位形成Int2-C8,经迁移插入过渡态TS1-C8得到远端烯烃配位的烯基铁物种Int3-C8;最终,Int3-C8与另一分子烯丙基锌试剂通过σ-键复分解释放产物并再生活性物种Int1-C8(图6e)。
结论
南开大学朱守非教授课题组以2-亚氨喹啉铁配合物为催化剂,首次实现了端炔的顺式反马氏烯丙基锌化反应。该反应表现出很高的活性(转化数高达1900)与精准的选择性控制(区域及立体选择性均高达> 95:5),同时具备广泛的底物适用范围与优异的官能团耐受性,为复杂多取代1,4-二烯化合物的快速构筑提供了高效途径。该方法被成功应用于RNA聚合酶抑制剂ripostatin B等生物活性分子关键中间体合成,合成效率显著高于文献报道的方法。机理研究表明,阴离子型烯丙基零价铁物种是反应的活性物种,其独特的活化模式与高催化活性可有效规避其他副反应;而2-亚胺喹啉配体合适的空间位阻是实现反马氏选择性的关键。这一研究不仅填补了普通端炔反马氏烯丙基金属化的空白,也再次证明:廉价金属铁在经典有机反应的选择调控中拥有巨大潜力。
这一成果近日发表于《美国化学会志》(J. Am. Chem. Soc.)杂志,文章的第一作者是南开大学博士研究生李潞杰,南开大学有机新物质创造前沿科学中心为论文通讯单位。
文献信息
Iron-Catalyzed anti-Markovnikov Allylzincation of Terminal Alkynes. Lu-Jie Li, Peng He, Xin-Yu Zhang, and Shou-Fei Zhu*. J. Am. Chem. Soc. 2026, DOI: 10.1021/jacs.5c21100.
