


研究背景
近年来,电化学合成凭借其绿色可持续的显著优势,已成为有机合成领域极具发展前景的前沿技术。该反应条件温和且过程可控,为开发高选择性氧化与还原方法提供了重要支撑。在各类选择性氧化反应中,烷基芳烃苄位 C(sp3)–H键的高效转化,更是受到有机化学家的广泛关注。然而,当前电化学条件下苄位C(sp3)–H键氧化,产物多以过度氧化生成的醛、酮为主(图1A)。究其原因,烷基芳烃与相应苄醇产物中苄位C(sp3)–H键的键解离能 (BDE) 及氧化电位十分接近。与之形成鲜明对比的是,能够实现苄位C(sp3)–H 键直接电化学羟基化的高效方法仍十分匮乏。
南开大学化学学院、有机新物质创造前沿科学中心仇友爱研究员课题组聚焦于有机电化学的研究(Acc. Chem. Res.2025, 58, 113-129)。此前,仇友爱课题组发展了以六氟异丙醇 (HFIP) 辅助的苄位 C(sp3)–H键直接羟基化策略,其核心在于HFIP可通过氢键作用来稳定苄醇,降低芳环电子云密度,从而抑制产物进一步氧化,提升反应选择性(J. Am. Chem. Soc. 2025, 147, 23297-23307; 图1B)。尽管该体系取得了一定进展,但酮类副产物仍然难以完全抑制。因此,迫切需要发展全新的电化学策略,实现苄位 C(sp3)–H键直接、高效且专一的羟基化转化。
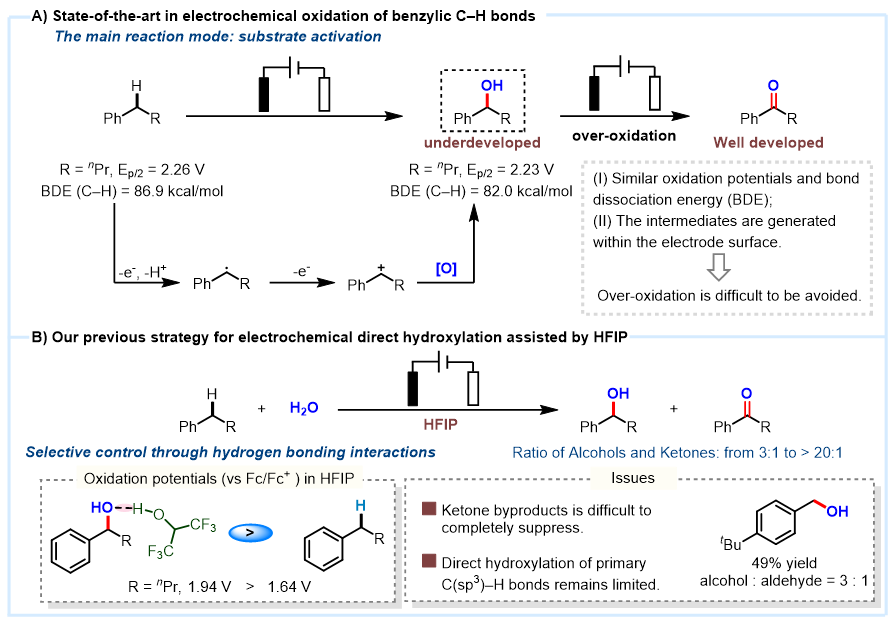
图1. 电化学苄位C(sp3)–H键的氧化策略
研究内容
基于该课题组前期水(氘水)参与的电化学转化的研究基础(近期工作如: J. Am. Chem. Soc. 2025, 147, 23297-23307; Angew. Chem. Int. Ed. 2025, 64, e202425634; Sci. Adv., 2026, 12, eaeb7677, Natl. Sci. Rev. 2026, 13, nwag047),同时受仿生铁催化剂选择性活化 C(sp3)–H 键的启发,仇友爱课题组提出了一种创新策略:通过水与仿生铁催化剂原位电生成高活性铁氧物种(Fe (IV)=O)替代底物直接电极氧化。接着与烷基芳烃发生电子转移,该过程可高效生成苄基自由基,随后经氧反弹机理精准构建苄醇产物。尤为关键的是,反应可在分离型“反应层中进行”,生成的苄醇产物不容易扩散至电极表面,从空间上阻断了过度氧化路径。近日,该课题组在Angew. Chem. Int. Ed.上发表论文,报道了电化学仿生铁催化策略,成功实现苄位C–H键高效、高选择性羟基化。该方法具有以下突出优势:a) 借助分离反应层与氧反弹机理,实现苄位C–H键专一羟基化;b) 广泛的底物范围,可兼容伯、仲、叔三类苄位C–H键;c) 可用于生物活性分子的后期修饰与合成,展现出良好的合成应用潜力;d) 成功实现了天然化合物的直接羟基化,并表现出优异的位点选择性,为复杂分子的修饰提供了新思路。
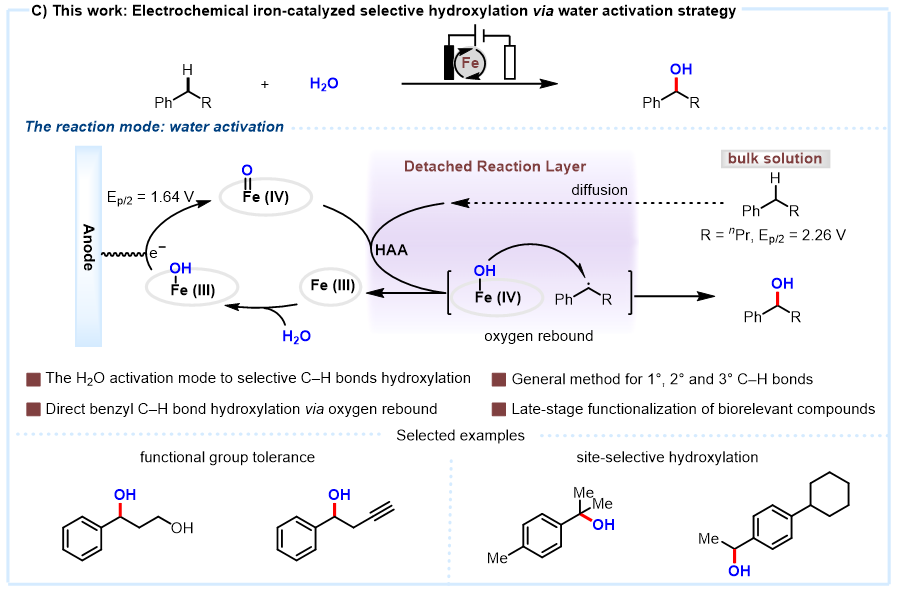
图2. 电化学仿生铁催化苄位C(sp3)–H 的羟基化反应
该方法的底物普适性良好,能够兼容1°、2°、3°三类苄位C–H键、杂环、以及含有不同官能团(如-Br、-OH、-CO2Me、C=C、C≡C等)的芳烷烃(图3)。此外,该策略还实现了一系列α-羟基酸酯的合成。
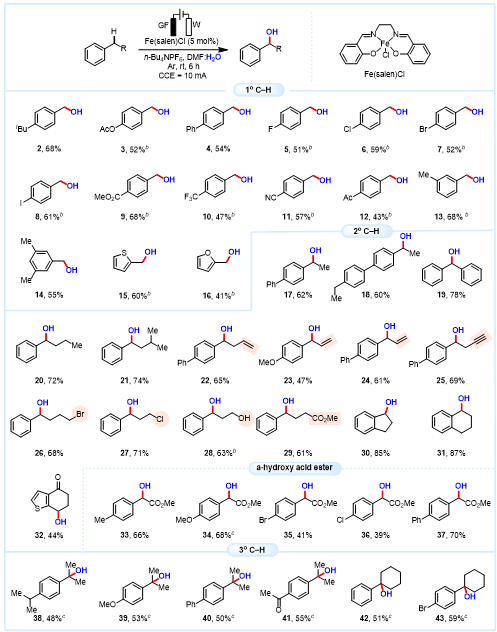
图3. 底物普适性研究
值得注意的是,该电化学合成策略在双苄位底物的转化中表现出优异的位点选择性(图4,上)。当底物同时含有1° 与2° 苄位 C–H 键时(44a−45a),羟基化反应优先发生在仲碳位点。类似地,在1° 与3° 苄位 C–H 键共存的底物中(46a),反应更倾向于在叔碳位点进行。这种显著的区域选择性可归因于中间体自由基的稳定性。除自由基稳定性外,空间位阻在此类选择性调控中同样起到关键作用。因此,对于同时含有仲、叔苄位 C–H 键的底物(47a−49a),羟基化反应依然优先选择位阻较小的仲碳位点。另外,该策略能够实现复杂天然产物、药物衍生物的后期羟基化修饰(图4)。以含有苄位C–H键的结构的药物(如非布司他、丙磺舒等)和天然化合物(如月桂酸、薄荷醇等)为底物时,均可在温和条件下进行羟基化反应,为药物研发和天然产物结构修饰提供了有效的手段(图4)。
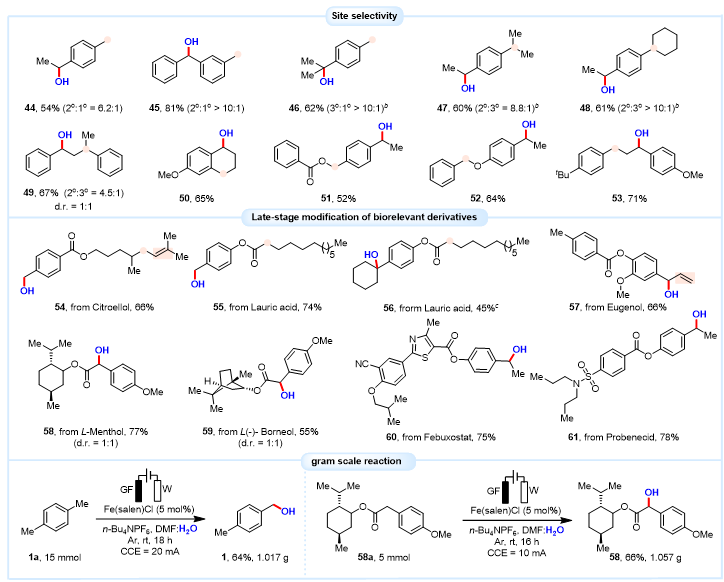
图4. 后修饰的天然产物普适性探索和克级反应
重要的是,该策略还可以拓展至天然产物与药物分子的直接羟基化反应(图5)。其中,雌酮与普罗雌烯均优先在3° 苄位C–H键上发生选择性羟基化。这一选择性源于对位给电子基团所引发的 p–π 共轭效应。有趣的是,洛索洛芬酯、布洛芬酯等的羟基化反应,均选择性发生在空间位阻较小的位点。此外,该电化学体系还可直接高效合成多种常见药物分子,如氯芬乙嗪、环扁桃酯等(图5)。
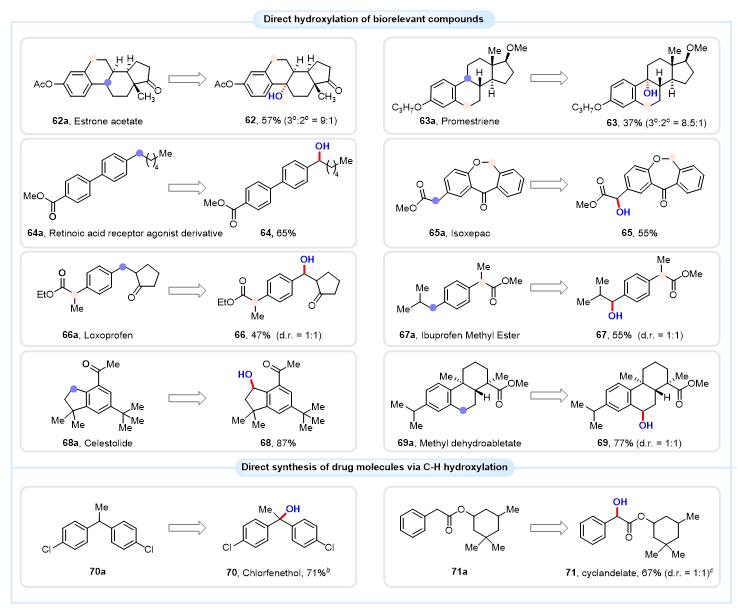
图5. 天然产物与药物分子的直接羟基化反应
接下来,作者对机理展开了研究。实验碳正离子捕获实验表明该反应不经历芳基自由基阳离子中间体的电子-质子 (ET-PT) 分步转移途径(图6A);动力学同位素效应 (KIE) 结果显示,氢原子攫取 (HAA) 过程可能为反应决速步(图6C)。循环伏安测试 (CV) 表明,证明了Fe(IV)=O 物种作为电荷传递介质,使均相电子转移主要发生在远离电极的反应层中,从而避免产物过度氧化;不同扫速下的 CV实验与旋转圆盘电极 (RDE) 实验进一步证实反应受动力学与扩散共同控制(图6D)。此外,紫外-可见吸收光谱监测发现(图6E),随电解时间延长,Fe(salen)Cl的特征吸收逐渐减弱并在6小时后趋于平稳,证明该催化剂在电解过程中持续发生结构与价态变化(如Fe(III) 向Fe(IV) 转化)并动态参与反应。
根据机理研究,作者提出了可能的机理循环(图6F)。首先,Fe(III)–OH 在阳极发生单电子转移氧化,生成高价活性中间体 Fe(IV)=O;随后该铁氧物种攫取底物烷基芳烃上的苄位氢原子,形成苄位自由基并再生为 Fe (IV)–OH;紧接着 Fe(IV)–OH 与苄位自由基发生氧反弹,完成羟基化并生成目标产物苄醇,同时再生Fe(III),实现催化循环。
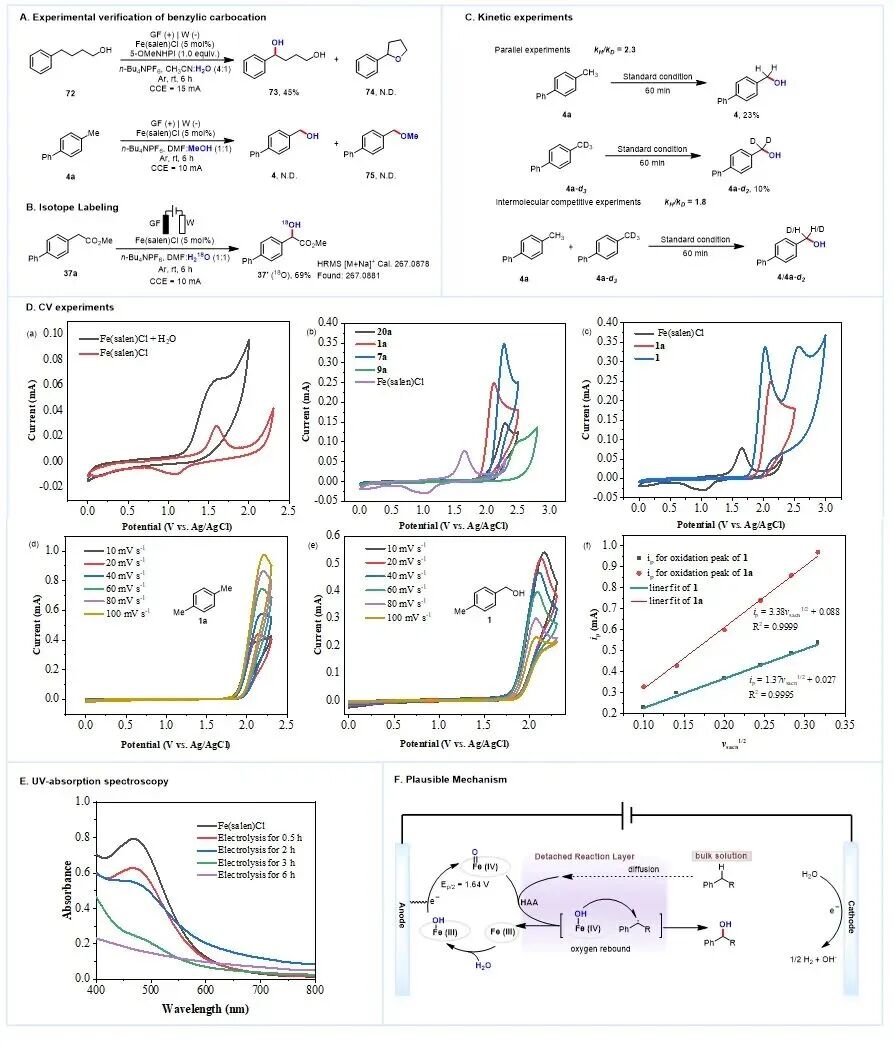
图6. 机理研究
小结
仇友爱团队发展了电化学仿生铁催化的苄位C–H键的直接羟基化反应,有效避免了电化学合成苄醇过程中常见的过度氧化问题。该方法底物适用性广,为构建含苄醇结构的高药用价值分子提供了高效途径。该工作得到了科技部重点研发专项、国家自然科学基金委、中央高校基本科研业务费及南开大学有机新物质创造前沿科学中心专项资金的支持。相关成果发表在Angewandte Chemie International Edition 上。南开大学化学学院、有机新物质创造前沿科学中心仇友爱研究员为通讯作者,博士生刘敏为文章的第一作者,南开大学有机新物质创造前沿科学中心为论文通讯单位。
文献信息
ElectrochemicalBiomimetic Iron Catalyzed Benzylic C–H Bonds Hydroxylation. Min Liu, Wentao Zhang, Cui Zhang, Chao Gao, Youai Qiu. Angew. Chem. Int. Ed., 2026, DOI: 10.1002/anie.9980873
来源:元素有机化学全国重点实验室
