


研究背景
立体专一的全碳四取代烯烃,广泛存在于天然产物及具有生物学或药学相关活性的分子中(图1A)。然而全碳四取代烯烃的立体专一性合成一直以来都是有机合成化学领域极具挑战的课题。传统的Wittig烯化反应、Peterson消除反应、烯烃复分解反应、Heck反应等经典合成方法,通常比较适合构建二取代和三取代烯烃,对于非环化四取代烯烃的合成存在着明显的局限性。此外,无论是通过炔烃的碳金属化、串联亲电捕获反应;还是多取代烯基底物参与的直接交叉偶联反应往往面临区域选择性或立体选择性控制难以控制、底物普适性差、且合成路线冗长等问题。该领域的挑战主要归咎于非环化全碳四取代烯烃双键具有高度位阻拥挤特性。一般来说,E-型异构体在热力学上更为稳定,但由于其与Z-型异构体之间的能量差异微乎其微,进一步增加了合成与构型控制的难度(图1B)。从反应的机理来看,原位新生成的多取代烯基-过渡金属络合物较易发生异构化,从而生成相应的Z/E-混合中间体,从而阻碍通过热力学调控获得具有合成应用价值的立体专一四取代烯烃(图1C)。尽管已有零星立体保持型四取代烯烃的合成方法,但通过立体反转交叉偶联策略从E-型烯基底物出发,通过立体翻转策略合成Z-型四取代烯烃的策略尚未实现。这一研究空白领域为突破全碳四取代Z-型烯烃这一合成难题提供了新的机遇(图1D)。
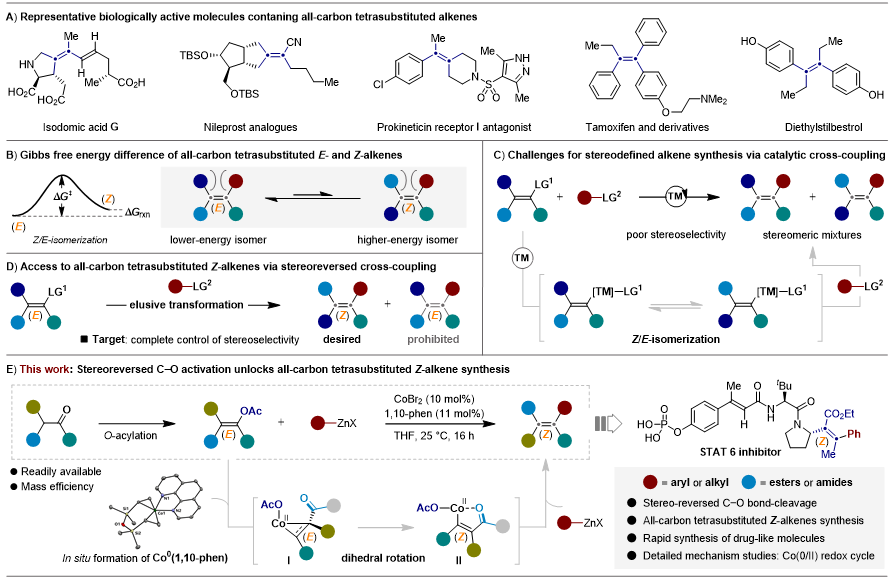
图1.全碳四取代烯烃立体选择性合成的研究进展。
研究内容
近日,南开大学化学学院、有机新物质创造前沿科学中心莫贞波研究员联合苏州大学李杰教授、武汉大学戚孝天教授开发一种实用的催化体系,通过立体反转的C−O键活化策略,选择性地实现有机锌试剂与多取代E型烯基乙酸酯之间的Negishi交叉偶联反应。通过简洁的制备路线进一步实现高价值生物活性分子的立体选择性构建,也证实了本方法在药物化学领域的巨大潜力(图1E)。
团队首先以E-烯基乙酸酯2a与芳基锌试剂1a为模型底物,对钴催化E-烯基乙酸酯立体翻转C−O芳基化反应进行了配体筛选(图2A)。研究发现,在催化量的CoBr₂存在下,以1,10-菲咯啉(L1)为配体,可以高效实现E-烯基乙酸酯2a立体反转C−O芳基化反应,以91%的产率和30:1的Z/E比得到四取代烯烃3(图2A)。值得注意的是,为了进一步探究配阴离子调控策略对锌试剂反应性的影响,团队制备了包括传统卤素配位的锌试剂在内的六种芳基锌试剂,并分别对其开展了平行实验。研究结果表明,与传统卤素配位的锌试剂相比,特戊酸(OPiv)配位的锌试剂在该反应体系中表现出了显著的反应优势(图2A,右)。
随后,团队对反应底物的适用范围进行了考察。值得注意的是,该催化体系展现出了非常广泛的底物普适性和官能团兼容性,各类高价值官能团,如卤素、噻吩、腈基、磺酰胺基等均可以在该反应体系中兼容,并以中等至良好的收率得到目标产物。其中,从相应的E-烯基乙酸酯到Z-选择性烯烃14的独特立体控制已通过单晶衍射进行了验证。含有Csp2−Br、Csp2−OTs或Csp2−OTf键的E-烯基乙酸酯亲电试剂能优先活化乙烯基Csp2−OAc键,而其它离去基团完全得以保持,以完全可控的化学选择性实现烯基C−O键的立体专一性官能化(图2B)。
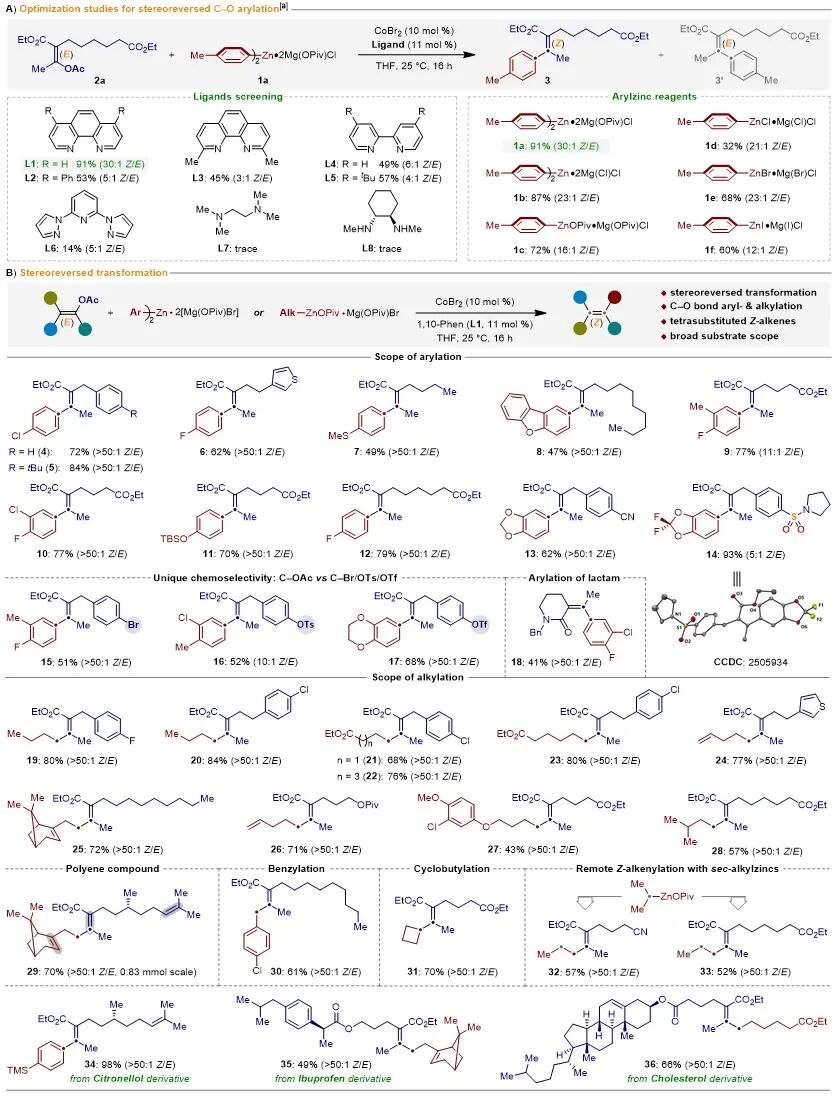
图2.通过立体反转的C−O键活化模块化合成Z型烯烃。
接着团队针对立体反转C−O键断裂实现的钴催化Negishi交叉偶联反应进行了详细的动力学研究。E-烯基乙酸酯(2a)和芳基锌试剂(1a)的浓度变化对初始反应速率几乎没有影响,而初始速率与钴催化剂浓度的关系图则呈现线性关系(图3A,i-iii)。因此,该钴催化的立体反转C−O芳基化反应对于烯基乙酸酯和有机锌试剂的浓度均为零级动力学,而对钴催化剂的浓度则为一级动力学。
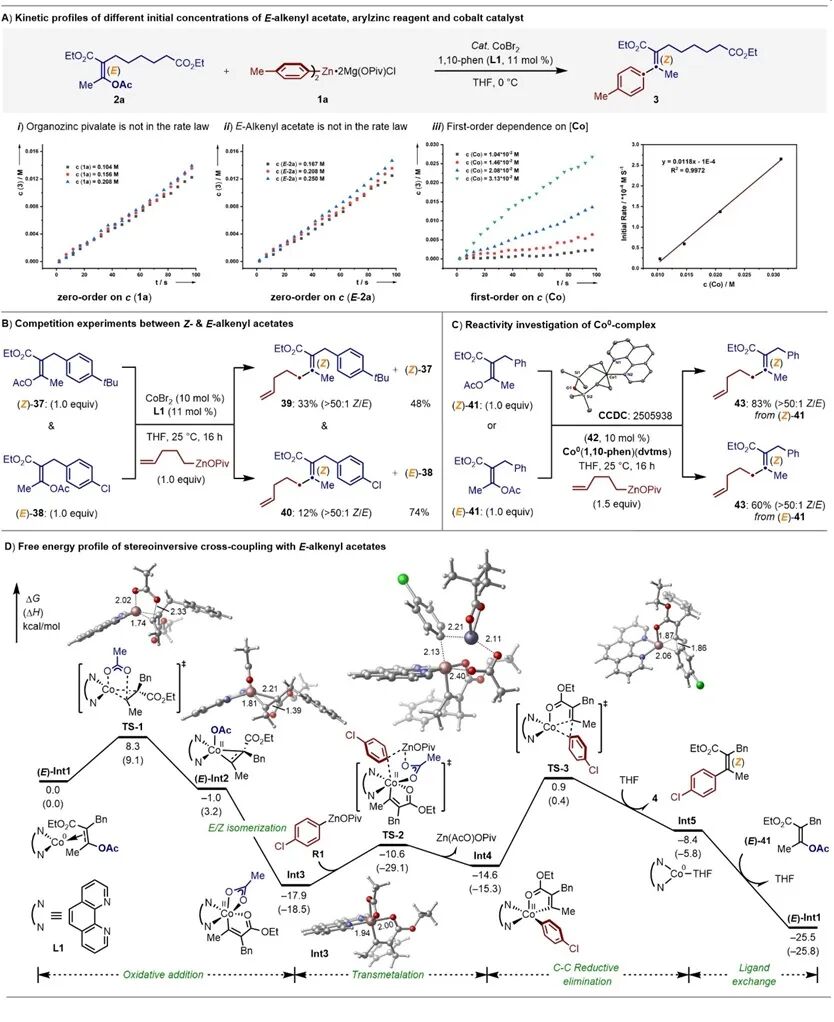
图3.机理研究。
接下来,Z-和E-烯基乙酸酯(37−38)之间的竞争实验表明,(Z)-37优先发生立体保持的偶联反应,以2.75:1的当量比生成Z-烯烃39和40。值得注意的是,回收的烯基乙酸酯的立体构型完全保持(图3B)。此外,为了研究催化活性钴配合物的结构,将KC8和二乙烯基四甲基二硅氧烷(dvtms)与CoIICl2(1,10-Phen)反应可以分离得到低价态Co0(1,10-Phen)(dvtms)物种42。进一步的单晶X-衍射明确了42的结构。值得注意的是,Co0(1,10-Phen)(dvtms)能够作为催化剂,高效地实现了Z-或E-烯基乙酸酯41的立体汇聚式C−O选择性烷基化反应,并以立体选择性地得到全碳四取代Z-烯烃43(图3C)。这些发现有力地支持了Co0(1,10-Phen)作为催化活性物种启动该偶联转化。
此外,多种Z/E-立体异构体比例接近1:1的烯基乙酸酯,能够与芳基锌试剂和烷基锌试剂发生Negishi交叉偶联反应,以优异的Z-立体选择性(> 40:1)独特地生成全碳四取代烯烃,而并未观察到相应的E-立体异构体(图4A)。同样,多种四取代Z-烯基乙酸酯能够与有机锌试剂发生立体保持的C−O芳基化和烷基化反应(图4B)。此外,团队进一步展示了该方法能够模块化地向复杂生物活性分子中引入全碳四取代Z-烯烃骨架。
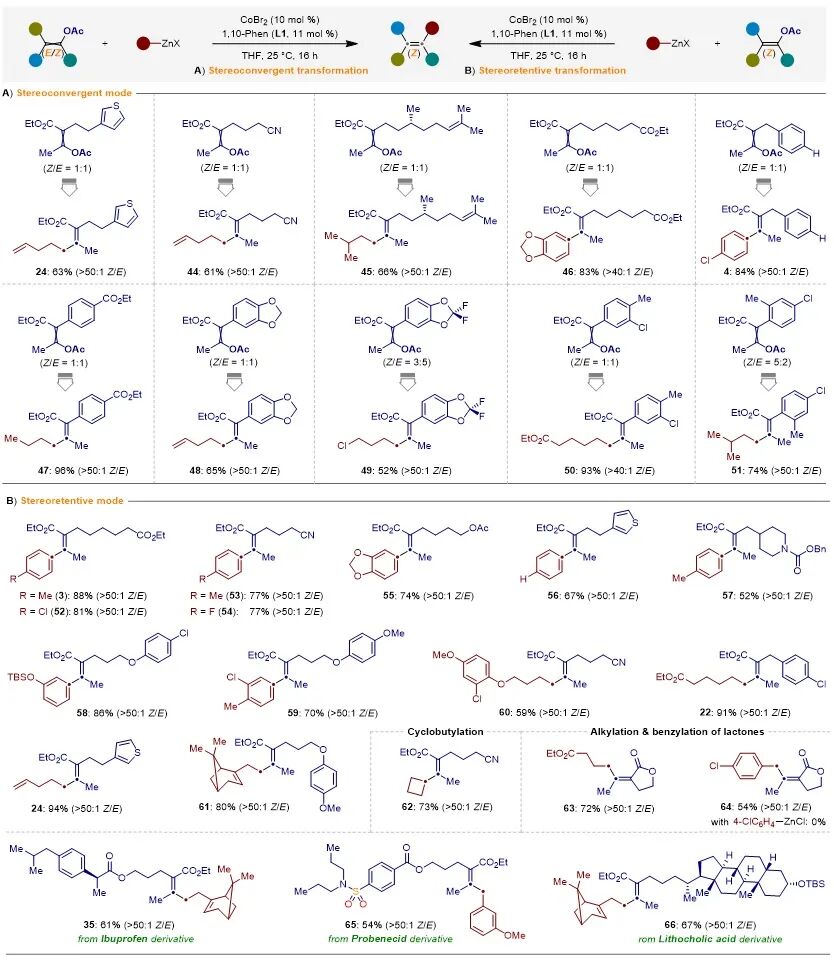
图4.底物范围。
为了进一步展示所得产物的合成应用价值,酯基可以转化为高价值的末端炔基结构,结合Click-反应得到三唑连接的Z-烯烃-药物的杂合体(图5A)。此外,基于立体汇聚式的C−O键活化构建全碳四取代Z-烯烃策略,可以成功合成获得前动力蛋白受体I拮抗剂的类似物(图5B)、信号转导和转录激活因子6抑制剂的类似物(图5C)、以及替马罗汀酸类似物(图5D)。这些合成应用极大地展示该合成策略的应用价值。
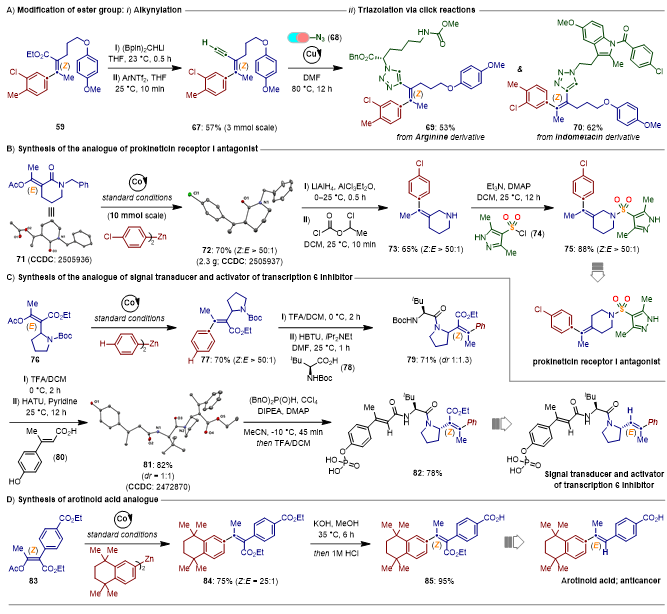
图5.合成应用。
总结
综上所述,团队报道了一种简单的钴催化体系,通过四取代E-烯基乙酸酯的C−O键活化实现立体反转Negishi交叉偶联反应。实现了Z-、E-烯基乙酸酯,以及任意比例的Z/E-混合物都能进行立体汇聚式的C−O转化,实现多取代烯基乙酸酯的立体选择性芳基化和烷基化反应。机理研究和理论计算研究表明,该钴催化的立体反转转化过程是通过Co0物种对C−O键的氧化加成启动,随后在酰基的辅助下发生二面角旋转,汇聚生成Z-构型的烯基CoII中间体,这是实现立体化学汇聚的关键。该反应由热力学驱动,其中还原消除被确定为决速步。这些研究加深了对钴的催化机制、以及立体选择性控制的理解,也展示了钴催化在立体汇聚式C−O键官能团化反应中的通用性。
相关研究以“Stereoreversed C−O Activation Unlocks All-Carbon Tetrasubstituted Z Alkene Synthesis”为题发表在《Journal of the American Chemical Society》上,苏州大学研究生陈开鑫、林杰和武汉大学彭佳丽为论文的共同第一作者,苏州大学李杰教授、武汉大学戚孝天教授、以及南开大学莫贞波研究员为通讯作者,南开大学有机新物质创造前沿科学中心为论文通讯单位。该研究得到了国家自然科学基金、江苏省自然科学基金、苏州市科技计划项目的经费支持。
文献信息
Stereoreversed C−O Activation Unlocks All-Carbon Tetrasubstituted Z Alkene Synthesis, Kaixin Chen, Jiali Peng, Jie Lin, Binjing Hu, Haihong Chen, Hongwei Jia, Changrui Nie, Zhenbo Mo*, Xiaotian Qi*, Jie Li*, J. Am. Chem. Soc., 2026, DOI: 10.1021/jacs.6c05845
来源:X-MOL资讯
