


研究背景
富锂锰基氧化物由于能够发生特定的阴离子氧化还原反应以提供高容量而备受关注,然而阴离子氧化还原的可逆性较差,导致严重晶格氧损失和表面结构演变。
2026年06月08日,南开大学化学学院、有机新物质创造前沿科学中心陈军院士、严振华副研究员团队在《Angewandte Chemie International Edition》期刊发表题为“Surface Chemical Disorder Engineering Enabled Superior Anion Redox for Li-Rich Mn-Based Cathode”的研究论文,团队成员张舒为论文第一作者,陈军院士、严振华副研究员为论文共同通讯作者,南开大学有机新物质创造前沿科学中心为论文通讯单位。
研究内容
该研究报道了一种通过将基于化学无序晶体织构整合到正极表面来解决这些问题的方案,该方案涉及通过阳离子在晶格中的过饱和占据来实现晶格氧的空间重排。这使得氧的电子结构离域化和多样化,增强了金属-氧轨道杂化,并有效提高了阴离子氧化还原反应的可逆性和动力学。同时,稳固的表面结构有效抑制了表面有害相演变以及电极/电解质界面副反应,维持了电化学过程的结构完整性。因此,所设计的改性正极展示了卓越的容量(291.8 mAh g⁻¹)、优异的长期循环稳定性以及电压保持能力(300次循环后容量保持率为90.5%,电压衰减为0.68 mV/cycle)。该研究揭示了表面化学无序以及过渡金属与氧之间强关联化学环境的作用,有望为正极材料的结构设计提供一种新范式。
锂离子电池(LIBs)在便携式电子设备和电动汽车中的广泛普及受其能量密度限制的制约,而能量密度主要由正极材料的性能决定。在诸多有前景的候选材料中,富锂锰基层状氧化物(化学式为xLi₂MnO₃·(1−x)LiTMO₂,TM=Mn、Ni、Co等)因其能够同时利用阳离子和阴离子氧化还原反应脱颖而出,从而提供极高的放电容量。然而,阴离子氧化还原的可逆性较差,通常会引发不可逆氧氧化,导致O−O二聚化并最终以O₂形式释放氧气。这一过程不仅会加速过渡金属迁移和结构重排(尤其是在表面),还会加剧与电解液的寄生反应,导致电极-电解液界面相的持续退化和不稳定演变。这些效应共同导致不均匀阴极电解质界面相形成以及Li⁺传输受阻,对富锂锰基正极的结构和界面稳定性提出了严峻挑战。因此,提高阴离子氧化还原反应的可逆性并稳定电极表面结构,是提升富锂锰基正极性能的关键。
先前用于增强阴离子氧化还原可逆性的策略主要集中于调控晶格氧的配位环境。代表性的方法包括元素取代(例如Mg²⁺、Nb⁵⁺、W⁶⁺、Be²⁺和高熵掺杂)、相控制(例如O2相)、孪晶结构以及超结构调控。尽管这些方法可以通过TMO₆八面体配位来微调电子结构并改善体相氧化还原可逆性,但它们在抑制界面氧活性以及减轻相关副反应方面的效果仍然有限。此外,可以通过引入外延涂层或引入具有高杨氏模量的第二相来构建表面保护层。外延涂层通常可以构建快速离子传输通道或形成HF隔离层以强化表面结构,而刚性基底通过增加晶格容忍度来抑制晶格畸变。然而,这些外源性涂层常常存在与主体材料晶格匹配度差、界面相容性弱的问题,在长期循环过程中带来应力积累和涂层剥落的风险。鉴于这些挑战,迫切需要一种能够同时促进可逆氧氧化还原并加固电极-电解质界面的本征晶体织构工程。
近期,短程化学无序——一种打破长程原子有序以产生局域化学异质性的晶体学构型——已成为调控电化学行为的一种有前景的途径。此外,基于经典晶体场理论,通过金属中心离子与配体之间的配位相互作用实现金属能级分裂和自旋态转变,这通常会引起晶体材料中阳离子的电子构型变化。因此,具有特定晶体场配位结构的过渡金属离子可能是短程化学无序的有效诱导剂。富锂锰基正极的失效机制被认为是晶格氧损失以及由阳离子从有序态向无序态转变引发的相变。在此,该研究开辟了一条新途径,将基于晶体织构的表面化学无序整合到Li₁.₂Mn₀.₅₄Ni₀.₁₃Co₀.₁₃O₂(LRM)正极中。研究人员选择了具有特殊电子构型的Fe和Ti元素来实现表面化学无序结构的构建。由于Fe³⁺(3d⁵)和Ti⁴⁺(3d⁰)的半满和全空电子构型,Fe³⁺和Ti⁴⁺元素的晶体场稳定能(CFSE)为0,这使其具有容纳外来电子的灵活结构能力,并易于形成无序阳离子结构。通过理论与实验相结合研究证明,层状结构主体通过引入额外的阳离子,能够自发诱导表面转变为无序畴区,有效形成原子尺度的保护屏障。离域氧电子结构显著增强了TM 3d-O 2p轨道杂化,从而有效锚定表面晶格氧并抑制有害的界面副反应。正如预期,借助稳固的表面阳离子无序畴区,所设计的改性正极展示了卓越的容量(291.8 mAh g⁻¹)、优异的长期循环稳定性(300次循环后容量保持率90.5%,电压保持率94.0%)。该研究揭示通过破坏主体晶体结构的化学有序来实现表面强化的方法,有望为正极材料的结构设计提供一种新范式。
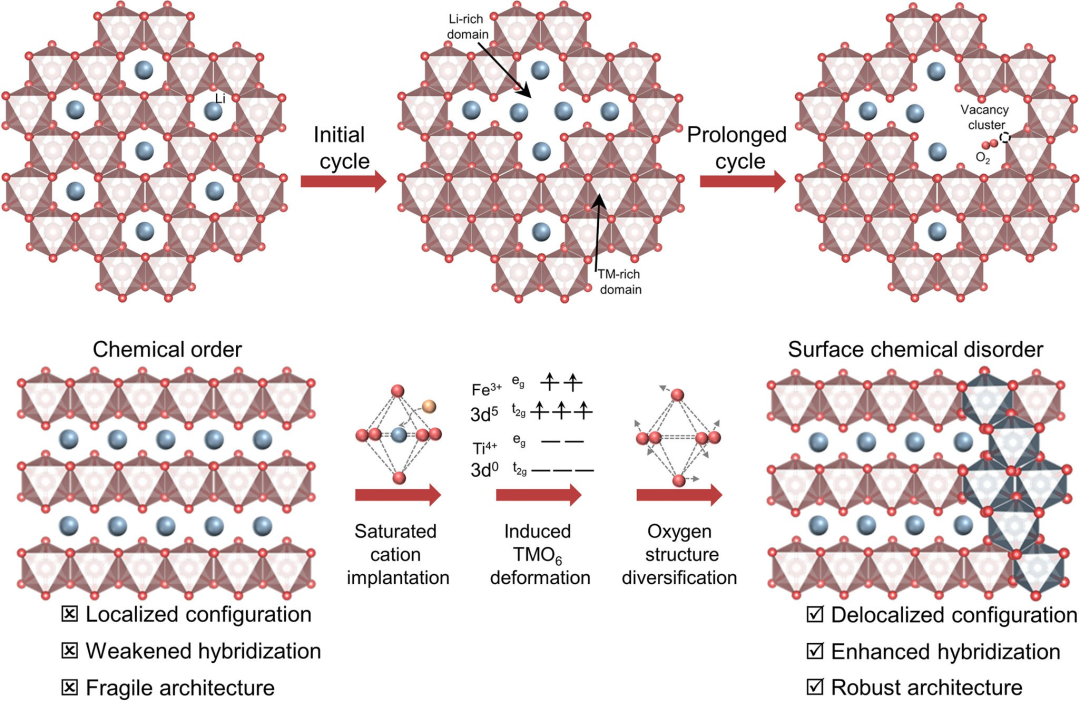
图1富锂锰基正极的失效机制与表面化学无序设计示意图。
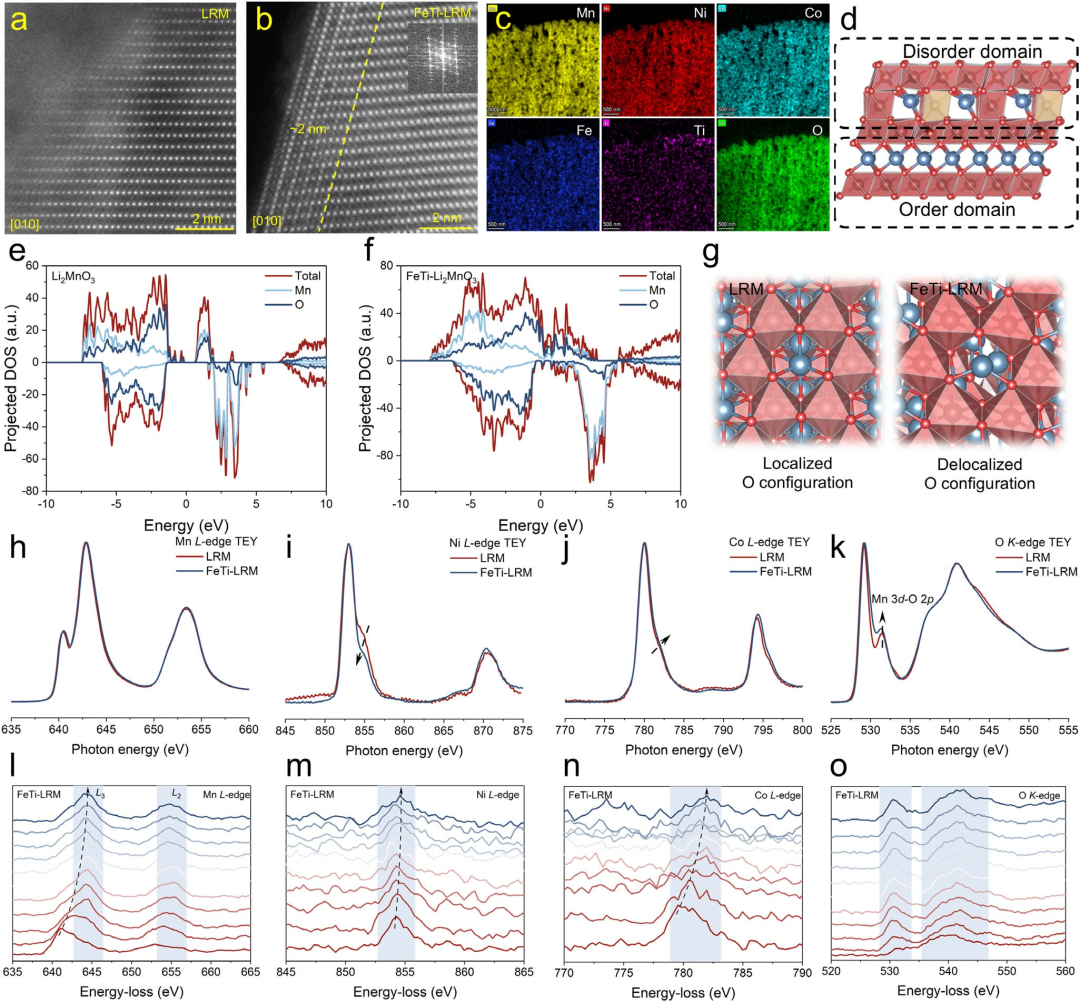
图2结构表征。LRM表面(a)和FeTi-LRM表面(b)沿[010]方向的高分辨HAADF-STEM图像。插图为相应的快速傅里叶变换图像。(c) FeTi-LRM中Mn、Ni、Co、Fe、Ti和O的EDS元素面分布图。(d) FeTi-LRM的晶体结构示意图。Li₂MnO₃(e)和FeTi-Li₂MnO₃(f)中Mn和O的计算投影电子态密度。(g) LRM和FeTi-LRM的LiMn₆结构单元的O构型图。LRM和FeTi-LRM的Mn L边(h)、Ni L边(i)、Co L边(j)和O K边(k)的XAS谱图。FeTi-LRM的Mn L边(l)、Ni L边(m)、Co L边(n)和O K边(o)的EELS谱图。从红色到蓝色,对应范围从表面0 nm到内部10 nm。
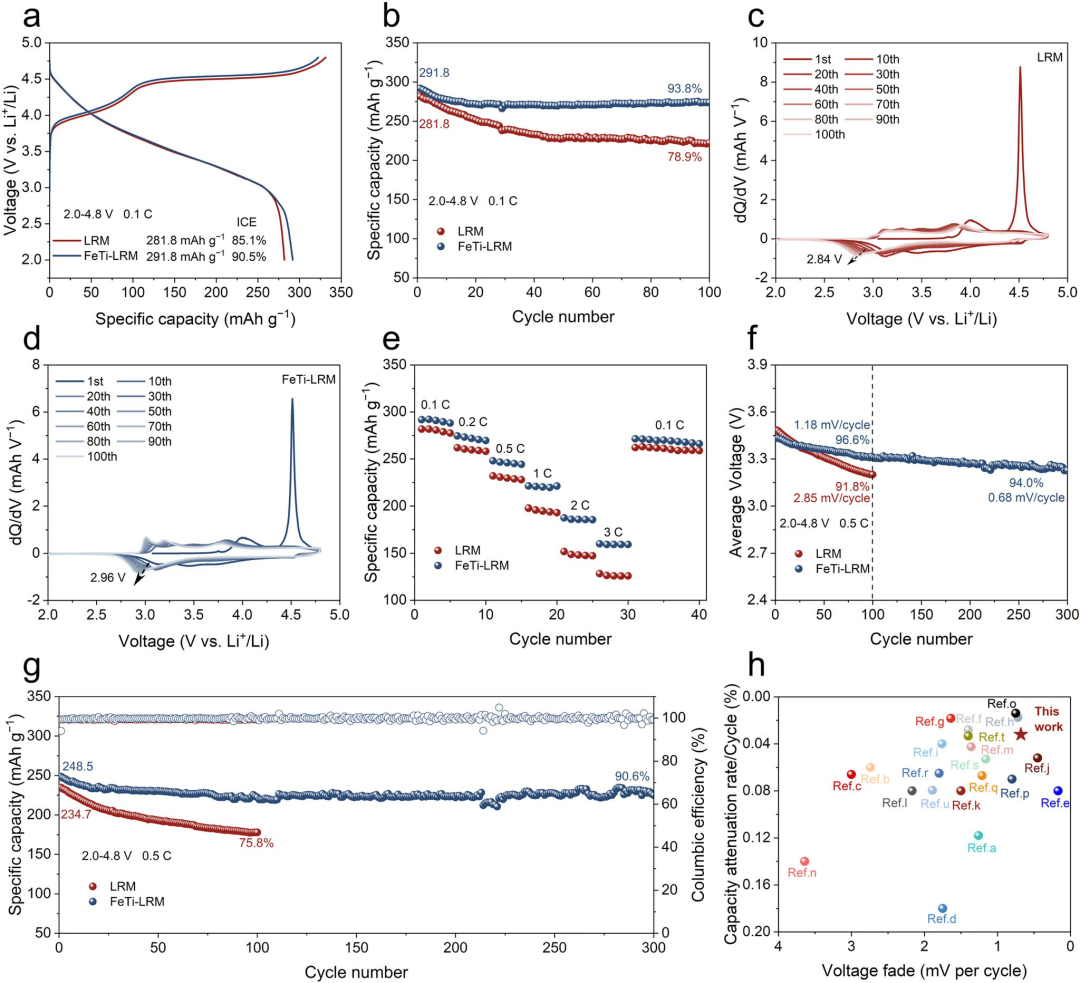
图3电化学性能。(a) 0.1 C下的恒流初始充放电曲线。(b) LRM和FeTi-LRM在0.1 C下的循环稳定性。LRM(c)和FeTi-LRM(d)在0.1 C循环过程中不同圈数对应的dQ/dV曲线。(e) LRM和FeTi-LRM的倍率性能。(f) LRM和FeTi-LRM在0.5 C下的平均电压。(g) LRM和FeTi-LRM在0.5 C下的循环性能。(h) FeTi-LRM与先前报道的LRM材料的综合性能对比。
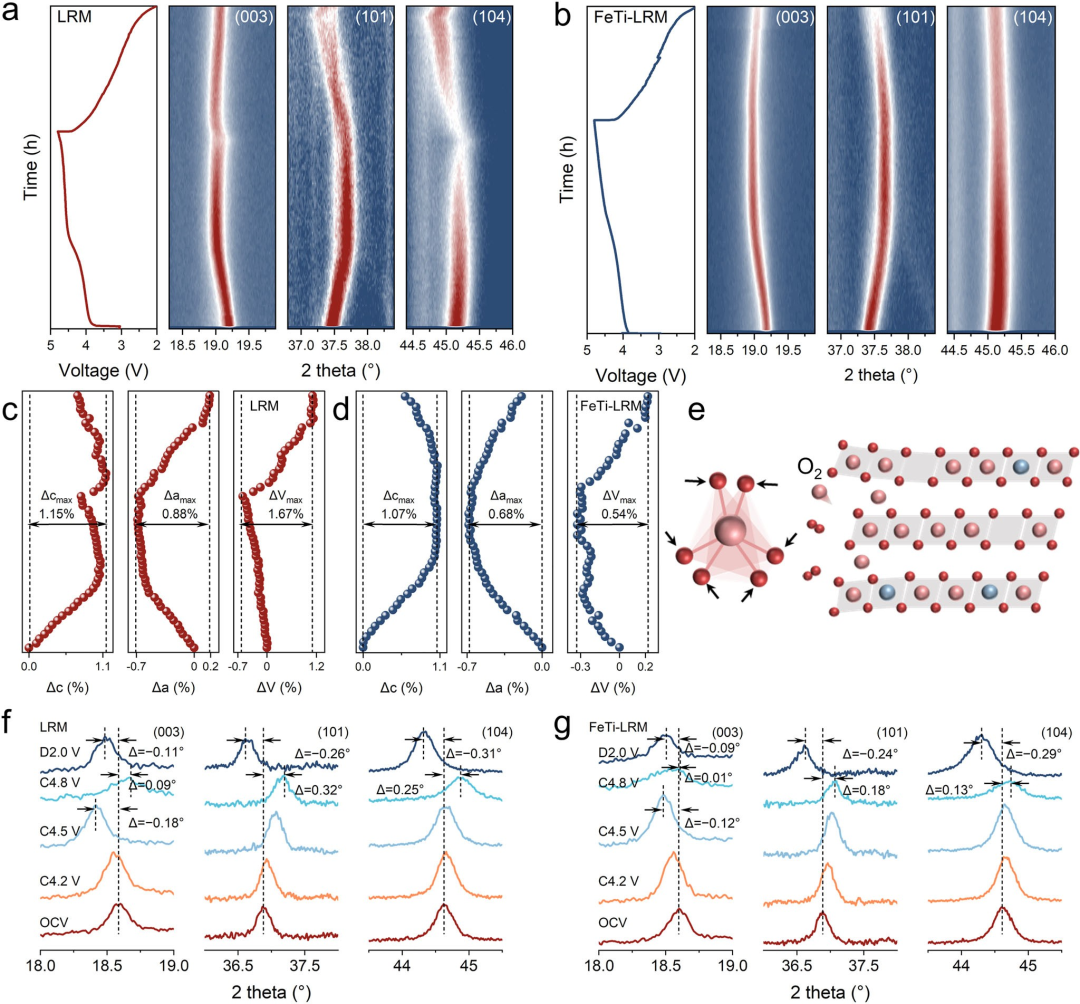
图4结构演变。LRM(a)和FeTi-LRM(b)的原位XRD图谱等高线图及相应的充放电曲线。通过将XRD数据与菱方R-3m空间群拟合得到的LRM(c)和FeTi-LRM(d)的原位XRD测试晶格参数(a、c和V)。(e) LRM的TMO₆畸变和晶格畸变示意图。LRM(f)和FeTi-LRM(g)的放大(003)、(101)和(104)衍射峰的XRD图谱演变。
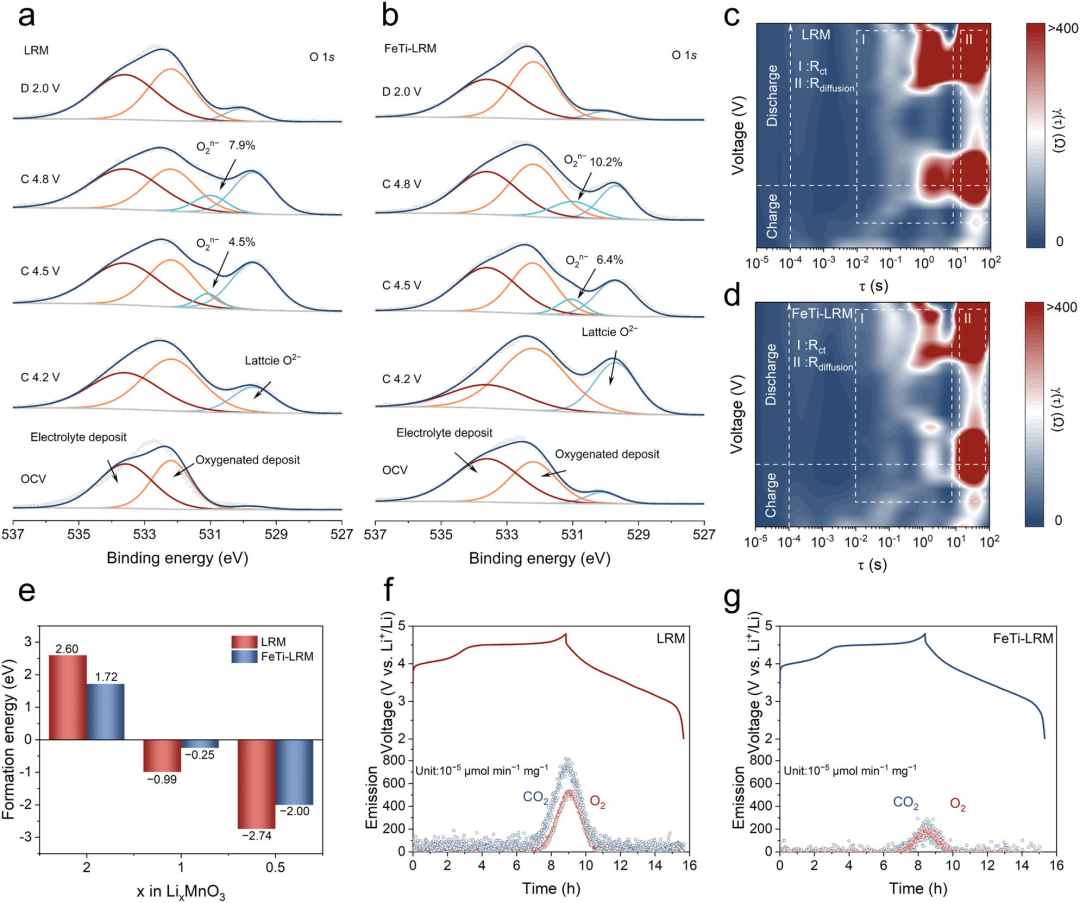
图5晶格氧演变。LRM(a)和FeTi-LRM(b)在不同电压状态下的XPS谱图演变。初始循环期间来自原位EIS谱图的LRM(c)和FeTi-LRM(d)的DRT分析。(e) LRM和FeTi-LRM在LiₓMnO₃中氧空位的形成能。通过原位DEMS测量得到的LRM(f)和FeTi-LRM(g)在初始循环期间的电压曲线及相应的气体(包括CO₂和O₂)释放。
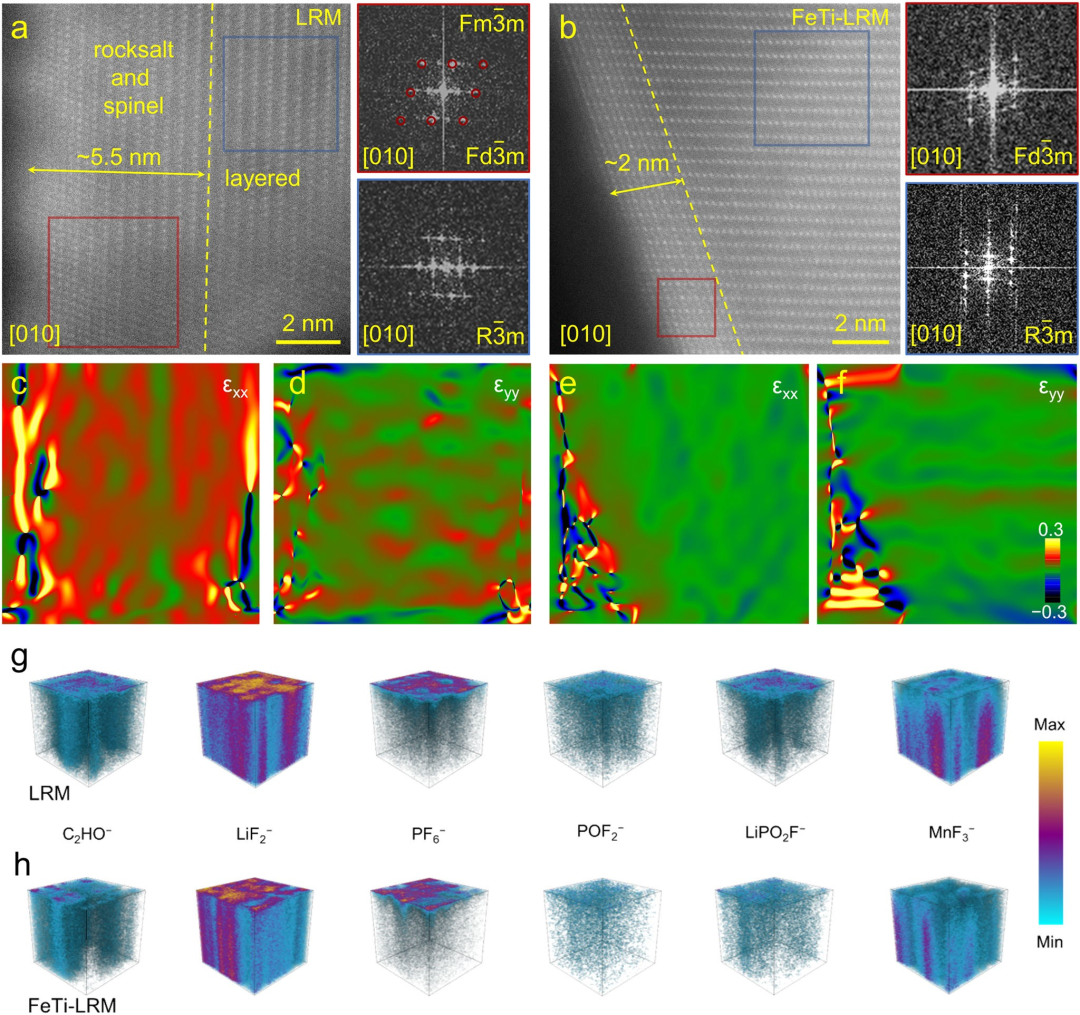
图6形貌与微观结构演变。循环100次后LRM(a)和FeTi-LRM(b)的HAADF-STEM图像及相应的FFT图像。通过GPA获得的循环后LRM(c-d)和FeTi-LRM(e-f)的应变状态。循环后LRM(g)和FeTi-LRM(h)的TOF-SIMS三维图像。
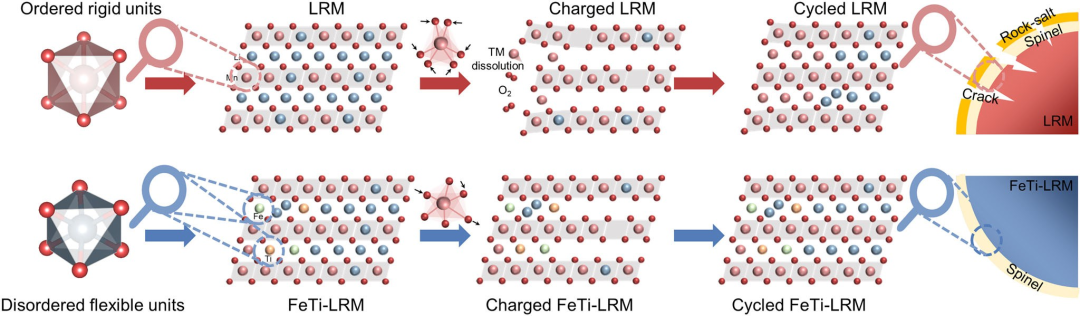
图7 LRM和FeTi-LRM的失效与改性机制示意图。
总结
综上所述,该研究展示了一种通过基于晶体织构工程构建表面化学无序畴区来强化富锂锰基氧化物正极的表面强化策略。额外过渡金属阳离子的引入成功诱导了从有序层状框架到部分无序表面层的结构转变。这种无序构型,借助FeO₆和TiO₆八面体独特的晶体场配位环境,显著增强了TM 3d与O 2p轨道之间的杂化。因此,在电化学循环过程中,相完整性和晶格氧稳定性得到显著改善,有效抑制了氧损失、过渡金属迁移和界面副反应。得益于这种稳固的表面结构,改性正极提供了291.8 mAh g⁻¹的高比容量,并表现出卓越的长期循环性能,在300次循环后仍保持90.5%的容量和94.0%的电压。该研究提出了一种反直觉方法,通过阳离子化学无序来稳定层状氧化物正极中的阴离子氧化还原活性,为依赖可逆氧还原反应的高能量密度电极材料的开发提供了有价值的设计原则。
来源:生化环材人
